# Virtual screening of the SARS-CoV-2 main protease (de.NBI-cloud, STFC)
Powered by:
Tim Dudgeon (opens new window), Simon Bray (opens new window), Gianmauro Cuccuru (opens new window), Björn Grüning (opens new window), Rachael Skyner (opens new window), Jack Scantlebury (opens new window), Susan Leung (opens new window), Frank von Delft (opens new window)
This repo serves as a companion to our recent docking simulations on the SARS-CoV-2 main protease.
It contains descriptions of workflows and exact versions of all software used. The goals of this study were to:
- Underscore the importance of access to raw data
- Demonstrate that existing community efforts in curation and deployment of computational chemistry software can reliably support rapid reproducible research during global crises
# Background
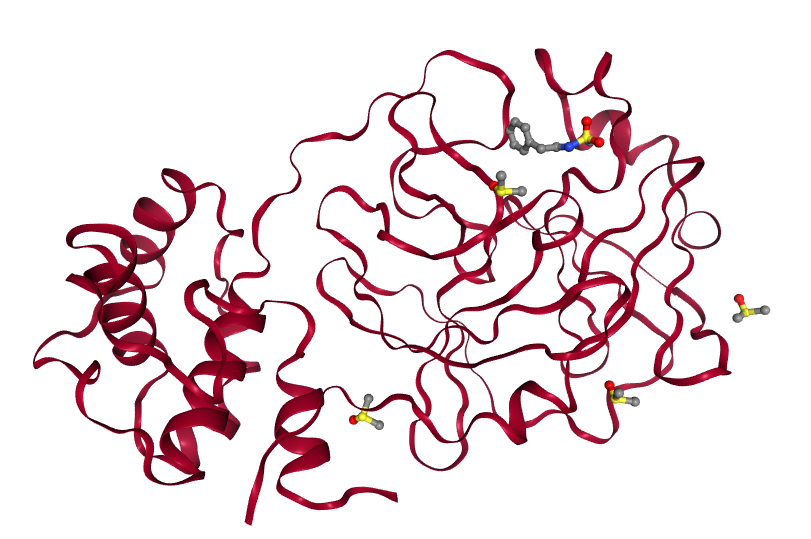
The Diamond Light Source's XChem team (opens new window) recently completed a successful fragment screen on the SARS-CoV-2 main protease (MPro) (opens new window), which provided crystal structures of the protein in complex with 55 different small molecules (fragment hits). These can be viewed interactively here (opens new window). In an effort to identify candidate molecules for binding, InformaticsMatters (opens new window), the XChem group and the European Galaxy team (opens new window) have joined forces to construct and execute a Galaxy workflow for performing and evaluating molecular docking on a massive scale, based on the set of fragment hits which are known experimentally to bind to the protein.
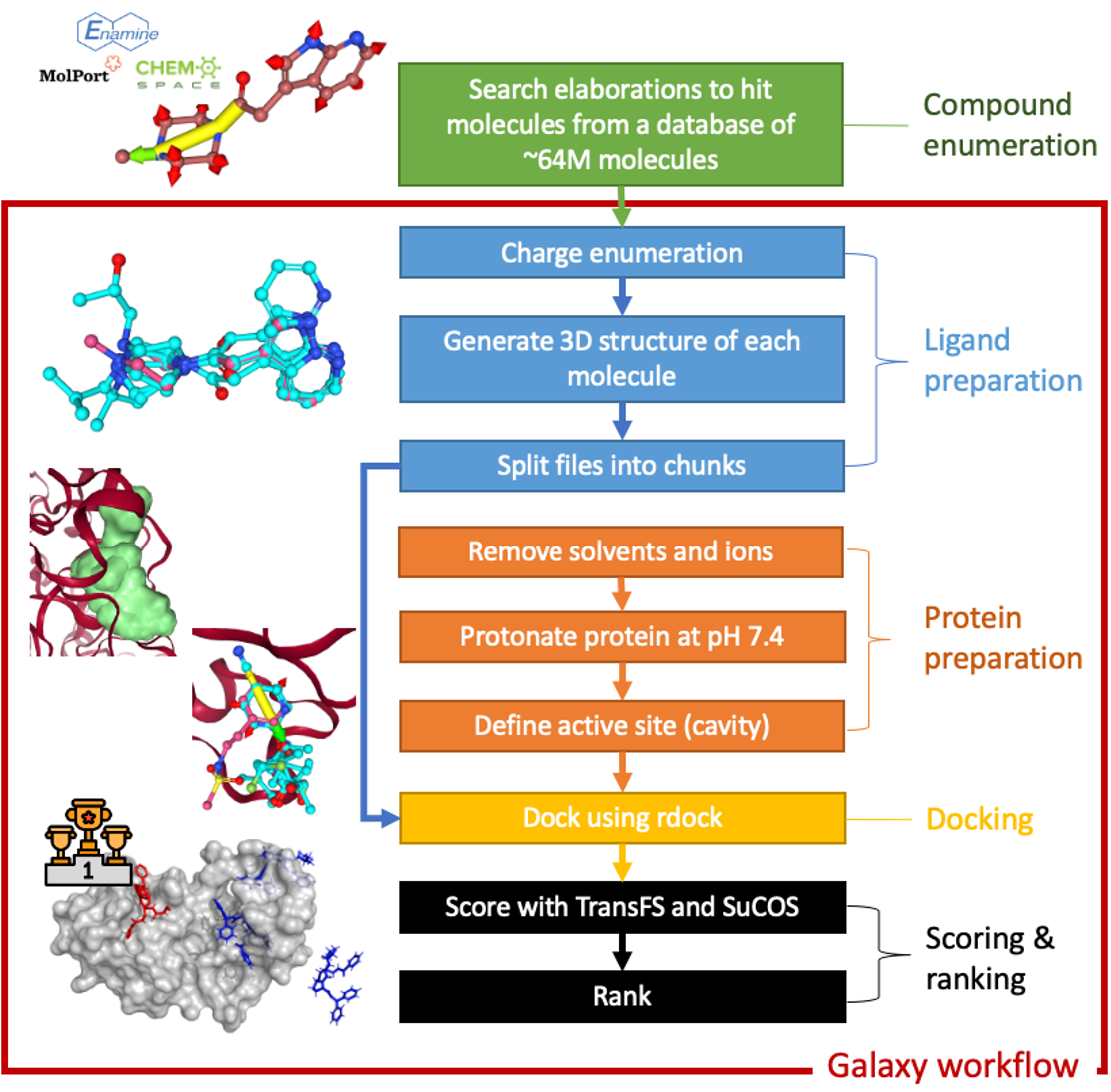
# Workflow
The diagram below describes the worfklow used in this work. Further details of the steps can be found in the compound enumeration and Docking and scoring workflow sections.
# Compound enumeration
An initial list of ~42,000 candidate molecules was assembled by using the Fragalysis fragment network (opens new window) to elaborate from the initial fragment hits. The fragment network takes a big set of compounds, and splits them up into parts – rings, linkers and substituents. These parts form the nodes in a graph network. The edges between these nodes describe how the bits of molecules can be linked together to make new molecules. From this information, we know how we can change a molecule by searching the network for new bits to add to an initial hit, with transformations described along the edges in the graph-network.
This was done using Informatics Matters’ Fragnet Search APIs (opens new window), querying a database of ~64M molecules available from Enamine REAL (opens new window), ChemSpace (opens new window) and MolPort (opens new window) using query parameters of 2 edge traversals and a change in heavy atom count of 5 and ring atom count of 2.
# Docking and scoring workflow (Galaxy)
The enumerated compounds were used as inputs for the docking and scoring workflow. The workflow consists of the following steps, each of which was carried out using tools installed on the European Galaxy server:
- Charge enumeration of those 42,000 candidate molecules to generate ~159,000 docking candidates.
- Generation of 3D conformations based on SMILES strings of the candidate molecules.
- Preparation of active site for docking using rDock.
- Docking of molecules into each of the MPro binding sites using rDock, generating 25 docking poses for each molecule.
- Evaluation of the docking poses using a deep learning approach (opens new window) developed at the University of Oxford, employing augmentation of training data with incorrectly docked ligands to prompt the model to learn from protein-ligand interactions. The algorithm was deployed on the European Galaxy server inside a Docker container, thanks to work by InformaticsMatters and the European Galaxy team.
- Scoring of the top scoring pose from each molecule against the original fragment screening hit ligands using the SuCOS MAX shape and feature overlay algorithm (opens new window), again deployed on the European Galaxy server by InformaticsMatters and the European Galaxy team.
- Selection of compounds available in the Enamine REAL (opens new window) and ChemSpace (opens new window) databases for purchase and further study.
This workflow was repeated for each of the 17 fragment screening crystal structures that were available at the time: Mpro -x1249, -x0072, -x0104, -x0107, -x0161, -x0195, -x0305, -x0354, -x0387, -x0434, -x0540, -x0678, -x0874, -x0946, -x0995, -x1077 and -x1093 (more hits have been found since).
Of these steps, the third (docking) is the most compute-intensive. Here, the project benefited from the enormous distributed compute capacity which underlies the European Galaxy project. Over 5000 CPUs were made available, provided by Diamond’s STFC-IRIS (opens new window) cluster at Harwell, UK and the de.NBI cloud (opens new window) in Freiburg, Germany. With each docking job requiring 1 CPU, thousands of poses could thus be docked in parallel, allowing millions of poses to be docked over a single weekend. The fourth step (pose scoring), while less computationally expensive, was accelerated thanks to GPUs provided by de.NBI and STFC. In total, the entire workflow described here took around 120,000 hours of CPU time (13 years) to complete.
All data is publicly available via https://usegalaxy.eu (opens new window), together with the workflows used for data generation, and we are working to provide more detailed documentation that will allow other users to perform similar studies, including on other systems. Histories for each fragment structures are provided here.
# Training
We have published a tutorial (opens new window), as part of the Galaxy Training Network (opens new window), describing how to perform a small-scale version of this analysis on the European Galaxy server. It also acts as a more detailed version of the documentation on this site.
# Galaxy workflow
| All in one virtual screening workflow |
|---|
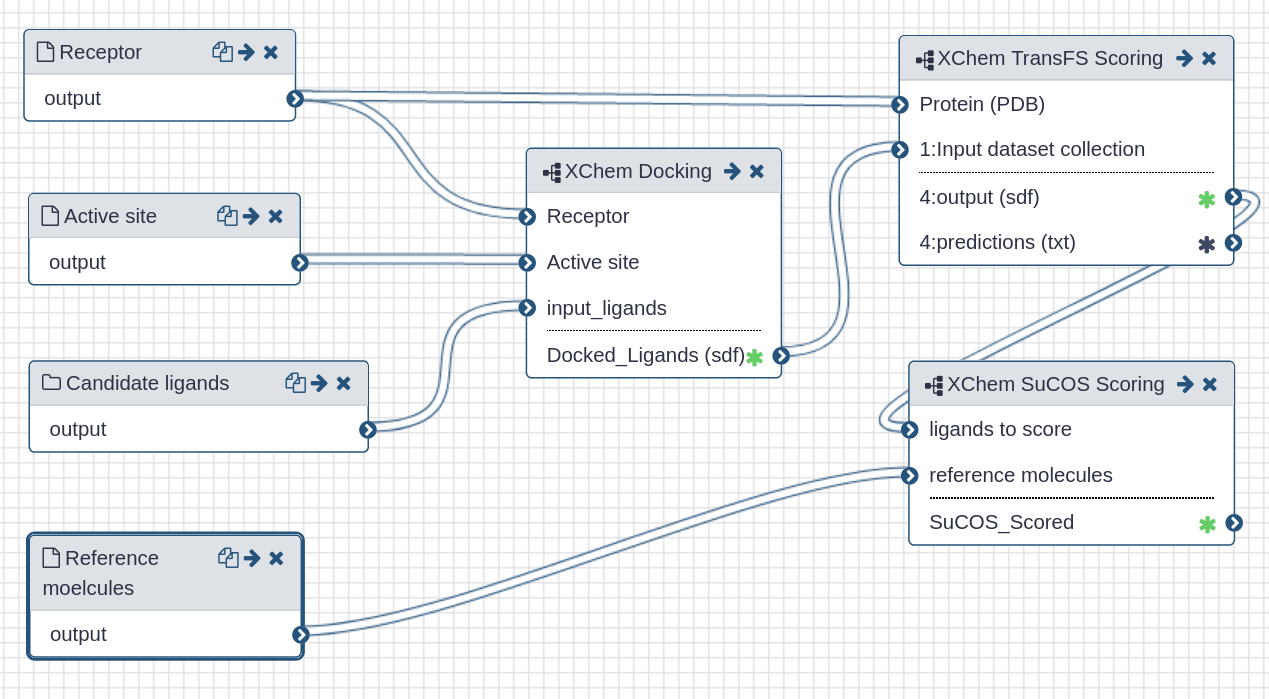 |
| This workflow contains all steps as sub-workflows. Please refer to the sub-pages for detailed information. |
# Future work
Having identified promising candidate ligands, we are now looking for funding to purchase compounds as a basis for further experimental study.
In addition we will be looking at newly released data here → Updates: Analysis of additional data
# Supporters and collaborators
The experiments have been performed using the Galaxy (opens new window) platform and open source tools from BioConda (opens new window) and conda-forge (opens new window). Tools were run using cloud resources provided by de.NBI (opens new window) and STFC (opens new window).


![]()


![]()
![]()
![]()
![]()
![]()
