Global platform for the analysis of SARS-CoV-2 data: Genomics, Cheminformatics, and Proteomics
Using open source tools and public cyberinfrastructure for transparent, reproducible analyses of viral datasets.
Powered by:
Visit our new SARS-CoV-2 surveillance site!
As of January 2022 this site will no longer be updated. Please visit our new site at https://galaxyproject.org/projects/covid19/ for the latest information on our surveillance efforts!The goal of this resource is to provide publicly accessible infrastructure and workflows for SARS-CoV-2 data analyses. We currently feature three different types of analyses: Genomics, Cheminformatics, and Proteomics.
Genomics
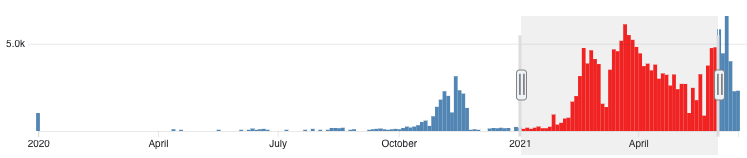
Freely accessible ready to use global infrastructure for SARS-CoV-2 monitoring new [bioRxiv]
Our system is specifically designed to encourage collaborative worldwide genomic surveillance to
rapidly identify and respond to emerging variants, its use of raw read data rather than
assembled genomes goes a step beyond current surveillance efforts. Specifically it enables the
coordinated worldwide surveillance of intra-patient minor AV frequencies: a distinction that
could yield decisive early-warnings of epidemiological conditions conducive to the emergence of
variants with altered pathogenicity, vaccine sensitivity or resistance.
BioRxiv Preprint
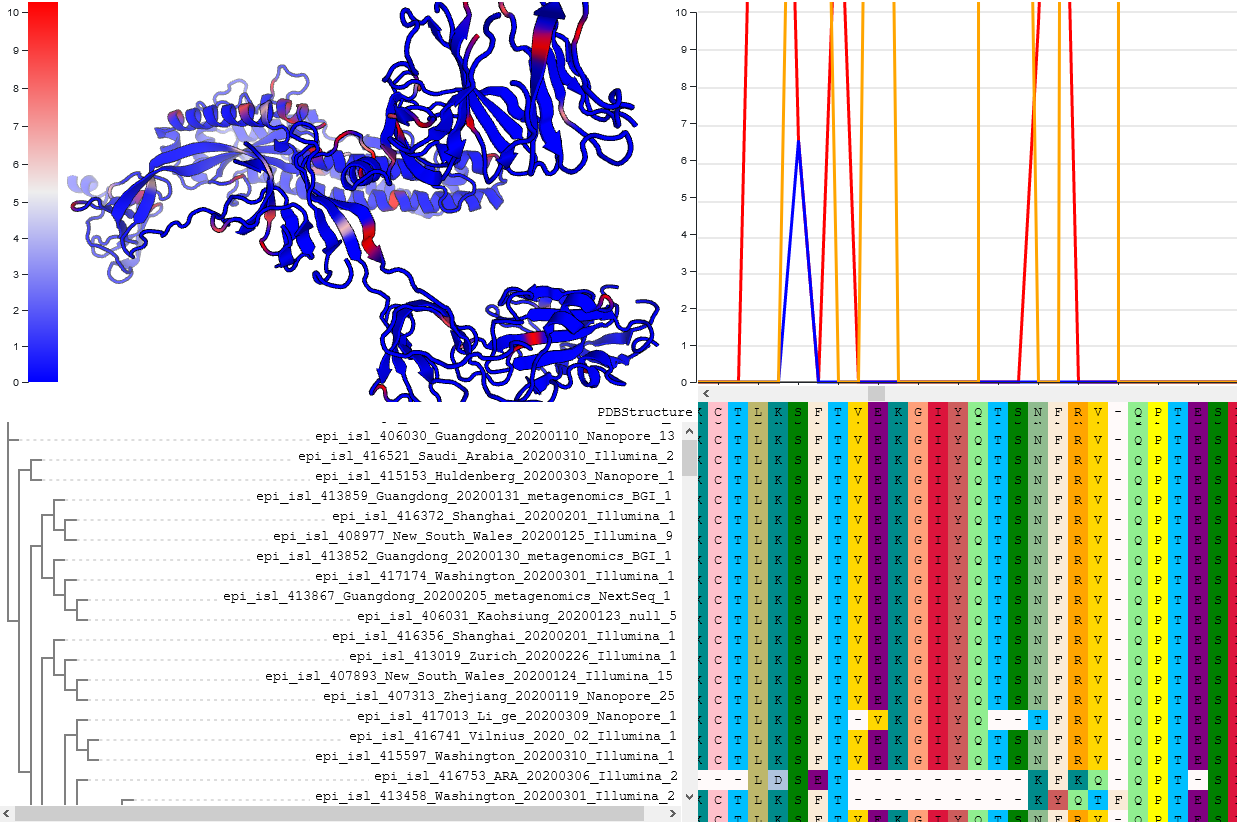
Evolution
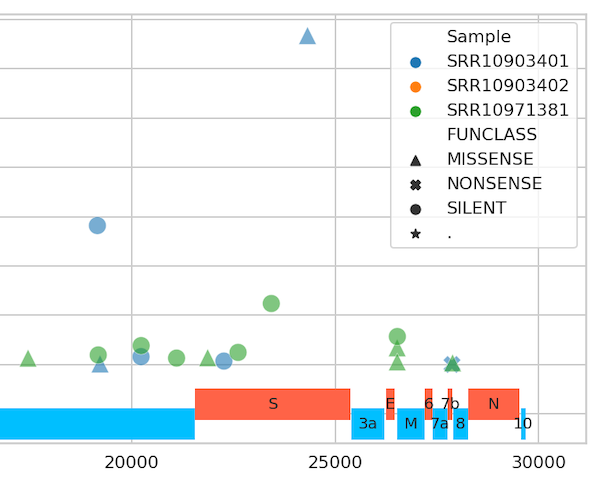
The list of selected sites in SARS-CoV-2 genome. It is continuously updated by the DataMonkey team using GISAID data as it accumulates. This list included all codons identified with FEL and MEME methods to be evolving under positive or negative selection.
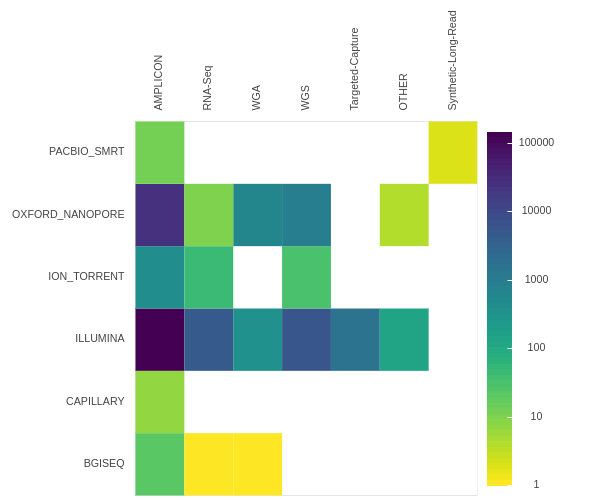
ARTIC: Amplicon based data analysis
A series of state-of-the-art workflows for the analysis of SARS-CoV-2 ARTIC data using the Galaxy platform.

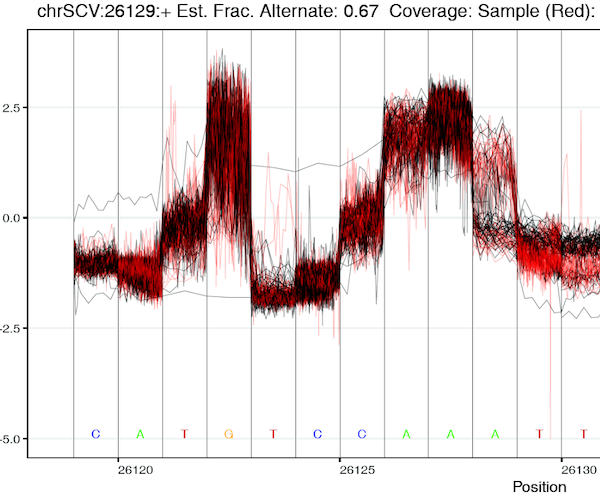
Analysis of SARS-CoV-2 transcription using Oxford Nanopore direct RNA sequencing [bioRxiv]
We use direct RNA sequencing to precisely annotate the open reading frames and the landscape of SARS-CoV-2 RNA modifications. Conservation of the RNA modification pattern during progression of the current pandemic suggests that this pattern is likely essential for the life cycle of SARS-CoV-2 and represents a possible target for drug interventions. We also provide Galaxy workflows for the analysis of DRS data.
No more business as usual - Agile and effective responses to emerging pathogen threats require open data and open analytics [PLoS Pathogens]
In an age of digital connectedness, open, highly accessible, globally shared data and analysis platforms have the potential to transform the way biomedical research is done, opening the way to “global research markets” in which competition arises from deriving understanding rather than access to samples and data.
Cheminformatics
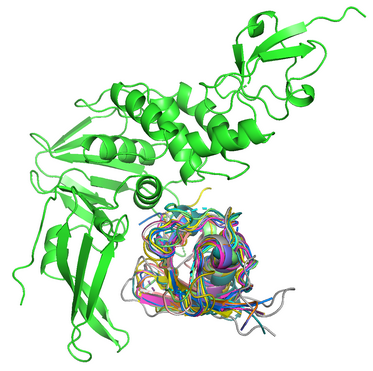
Fast and accurate genome-wide predictions and structural modeling of protein-protein interactions using Galaxy new
Identifying interacting proteins reveals insight into living organisms and yields novel drug targets for disease treatment. This is a publicly available, automated pipeline to predict genome-wide protein-protein interactions and produce high-quality multimeric structural models.
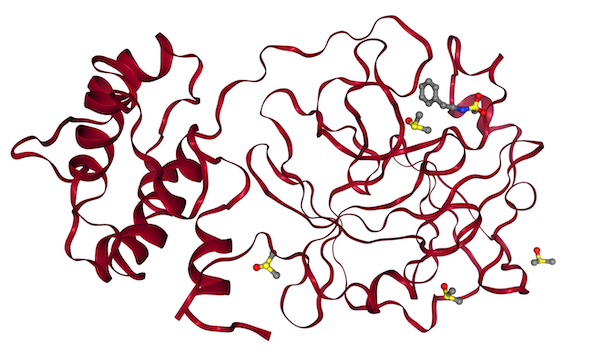
Docking simulations on the SARS-CoV-2 main protease.
A Galaxy workflow for performing and evaluating molecular docking on a massive scale, based on the set of fragment hits which are known experimentally to bind to the protein.
Proteomics
Galaxy workflows for analysis of Mass Spectrometry datasets from cell culture samples, clinical samples and protein-protein interaction datasets as well as functionality for performing metaproteomics analyses.

The analyses have been performed using the Galaxy (opens new window) platform and open source tools from BioConda (opens new window). Tools were run using XSEDE (opens new window) resources maintained by the Texas Advanced Computing Center (TACC (opens new window)), Pittsburgh Supercomputing Center (PSC (opens new window)), and Indiana University (opens new window) in the U.S., de.NBI (opens new window), VSC (opens new window) cloud resources and IFB (opens new window) cluster resources on the European side, STFC-IRIS (opens new window) at the Diamond Light Source, and ARDC (opens new window) cloud resources in Australia.



![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()


![]()




![]()
![]()