# Live Resources
| usegalaxy.eu | usegalaxy.org.au |
|---|---|
# Background
The ARTIC Network (opens new window) is a collaborative project between a number of laboratories around the world that wants to provide protocols and advice on viral outbreaks. Their website states that part of their goal is to develop "an end-to-end system for processing samples from viral outbreaks to generate real-time epidemiological information that is interpretable and actionable by public health bodies."
The ARTIC network has made available laboratory protocols, primer sets and bioinformatics workflows. The protocol they use relies on targeted amplification of viral sequences using tiled, multiplexed primers, an approach first described for Zika virus sequencing (opens new window).
Since this approach, as the ARTIC website states, "has been proven to have high sensitivity and work directly from clinical samples (compared to metagenomics approaches)", it has been widely adopted by groups worldwide and a fair percentage of the COVID genome sequences uploaded to GISAID and raw reads uploaded to the SRA have been produced by this method.
Along with the laboratory protocols for sequencing (both for Illumina and Nanopore sequencing technologies), several tools and workflows have been developed to handle what is essentially non-random sequencing data.
# Analyzing ARTIC data with Galaxy
# What's the point?
Our goal is to enable and provide state-of-the-art workflows for the analysis of SARS-CoV-2 ARTIC data using the Galaxy platform.
While it is possible to treat ARTIC data like regular whole-genome sequencing (WGS) data and analyze it with WGS workflows like the ones we developed for the analysis of within-samples variation, failure to take the specific nature of the data, and specifically the presence of amplicon primer sequences, into account would lead to suboptimal results.
# A Galaxy workflow for the analysis of Illumina paired-end sequenced ARTIC amplicon data
Compared to our workflow for WGS data, the workflow optimized for ARTIC data analysis includes the following changes:
No effort is made to deduplicate the sequenced reads data
Identical pairs of reads are commonly seen with highly amplified data from narrow ampliconic regions, but cannot be seen as prove that the reads have originated from the same viral template molecule.
Amplicon primer sequences are trimmed off the sequenced reads before variant calling
A naive analysis including resequenced primer sequences in the variant calling step would underestimate the frequency of variant alleles at primer binding sites, which, in turn, may lead to not calling real variants at all or to misclassifying fixed genomic variants as within-sample variation.
We use the iVar software (opens new window) from the Andersen Lab at the Scripps Research Institute in California for trimming the primer section of reads after mapping based on known primer binding sites.
This step also reduces strand-bias for variants at primer binding sites.
Note also that Illumina-sequencing protocols for the ARTIC approach typically include random fragmentation of the amplicons as part of their library preparation step in order to achieve full coverage of all amplicons. To account for this, we do not use iVar's option to discard reads that do not end in a primer sequence.
Read pairs amplified with a potential bias in primer binding are not used for calculating variant call statistics
If within-sample variation affects a primer binding site, the primer is expected to bind preferentially to the perfectly matching reference sequence as opposed to sequences carrying the variant allele, i.e., amplicons generated with such biased primers will more likely capture the sequence of viral template molecules without the variant than with it, and this extends to any linked variants within the amplicon region.
In order to avoid an allele frequency bias for such linked variants, one solution is to not use data from biased amplicons (for which at least one primer binding site displays within-sample variation) at all.
We implement this idea and perform a first round of variant calling to detect primer binding site variants. We then use iVar again to remove reads from biased amplicons prior to a second round of unbiased variant calling.
Since removing the information from whole amplicons is a rather drastic measure, which will leave a gap of no coverage in the tiled layout of reads, we then compare the variant lists before and after amplicon removal, and add variants lost in the second round of calling back in to the final list of variants. Those variants get flagged as not having passed amplicon removal though to indicate that their call statistics might not be reliable.
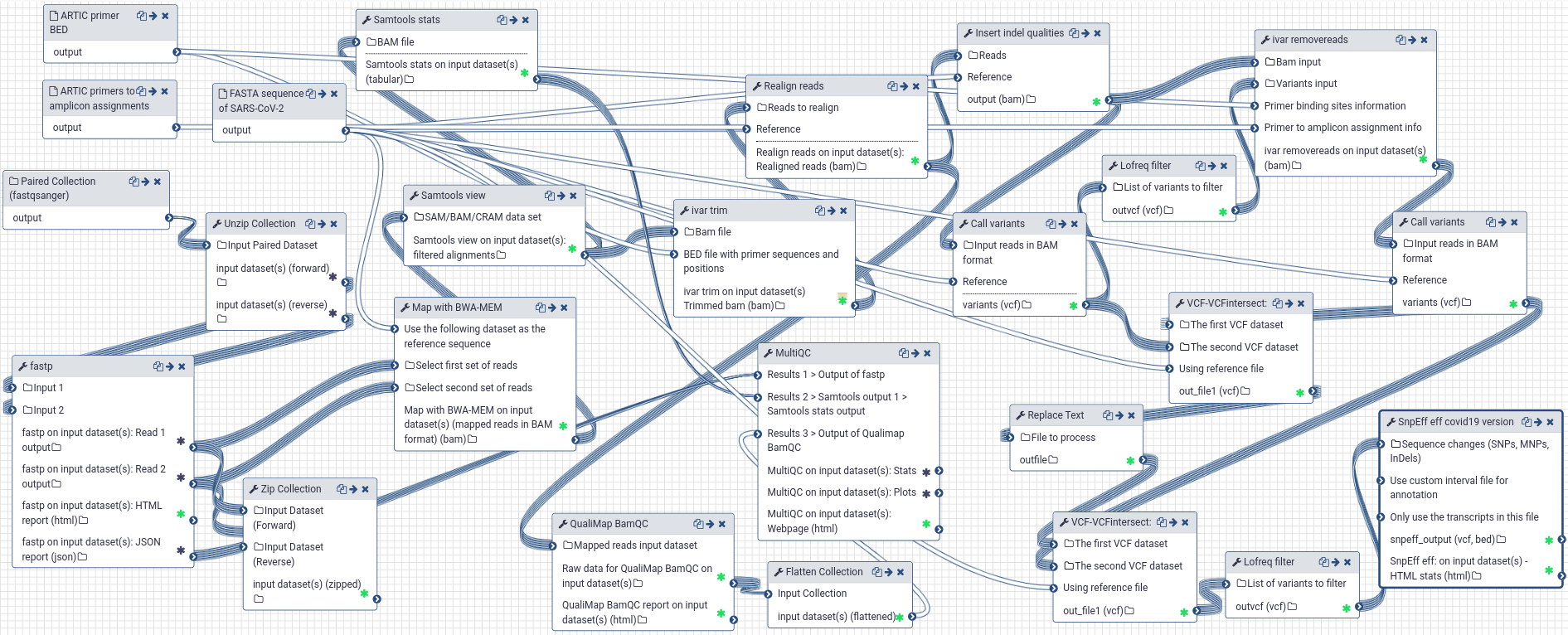 |
|---|
| Galaxy workflow for the analysis of Illumina paired-end sequenced ARTIC amplicon data |
# Required Inputs
The workflow requires:
A collection of the forward and reverse reads in
fastqformat of all samples to be analyzedThe SARS-CoV-2 reference FASTA sequence (opens new window)
A BED file describing the binding sites of all primers used to generate the tiled amplicons
The ARTIC project provides BED files for use with this workflow as part of their SARS-CoV-2 resources repo (opens new window).
For reproducibility we provide here copies of the:
Please take care to use the correct primer BED file corresponding to the version of the primers used during sequencing! If in doubt, consider this:
- Later BED file versions should also work reasonably well with older sequencing data, while the other way round is more problematic
- v2 of the primer scheme has been in use for only a very short time before it got superseded by v3.
An amplicon info file
This tabular file should consist of one line per amplicon, which should list all primers involved in the generation of this amplicon. For reproducibility we provide files with:
# Outputs
The workflow generates a collection of variant lists (one VCF dataset per sample).
Each dataset lists all variants discovered during variant calling and meeting minimal quality criteria.
The VCF FILTER column is used to indicate variants with an apparent allele frequency of < 0.05, variants failing a strand-bias test, and variants that could not be recalled upon removal of reads from biased amplicons.
![]()
![]()
![]()
![]()
# BioConda
Tools used in this analysis are also available from BioConda:
| Name | Link |
|---|---|
multiqc |  (opens new window) (opens new window) |
qualimap |  (opens new window) (opens new window) |
fastp |  (opens new window) (opens new window) |
bwa |  (opens new window) (opens new window) |
samtools |  (opens new window) (opens new window) |
ivar |  (opens new window) (opens new window) |
lofreq |  (opens new window) (opens new window) |
vcflib |  (opens new window) (opens new window) |
snpeff |  (opens new window) (opens new window) |
# Reference
Nathan D Grubaugh, Karthik Gangavarapu, Joshua Quick, Nathaniel L Matteson, Jaqueline Goes De Jesus, Bradley J Main, Amanda L Tan, Lauren M Paul, Doug E Brackney, Saran Grewal, Nikos Gurfield, Koen KA Van Rompay, Sharon Isern, Scott F Michael, Lark L Coffey, Nicholas J Loman, Kristian G Andersen, An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar (opens new window), Genome Biology 2019 20:8