# Proteomics analysis of SARS-CoV-2 infected cell culture samples
# Live Resources
| usegalaxy.eu |
|---|
# Description
Davidson et al (opens new window) used SARS-CoV-2 grown in Vero E6 cells to propagate the novel coronavirus and generate viral transcriptome and tandem mass spectrometry was used to investigate the proteome and phosphoproteome of the virally infected cells. The transcriptome was usedas an input for an ORF-centric pipeline.Their analysis detected a 24 nt in-frame deletion in mRNA encoding the spike (S) glycoprotein and was predicted to remove a proposed furin cleavage site from the S glycoprotein. Tandem mass spectrometry identified over 500 viral peptides and 44 phosphopeptides in virus-infected cells, covering almost all proteins predicted to be encoded by the SARS-CoV-2 genome, including peptides unique to the deleted variant of the S glycoprotein. Mass spectrometry data was acquired on an Orbitrap Fusion Lumos mass spectrometer (Thermo Scientific) in data-dependent acquisition mode and deposited in ProteomeXchange repository (PXD018241). We were interested in using the mass spectrometry dataset and searching it against the viral proteome to detect viral peptides from these samples.
# Workflow
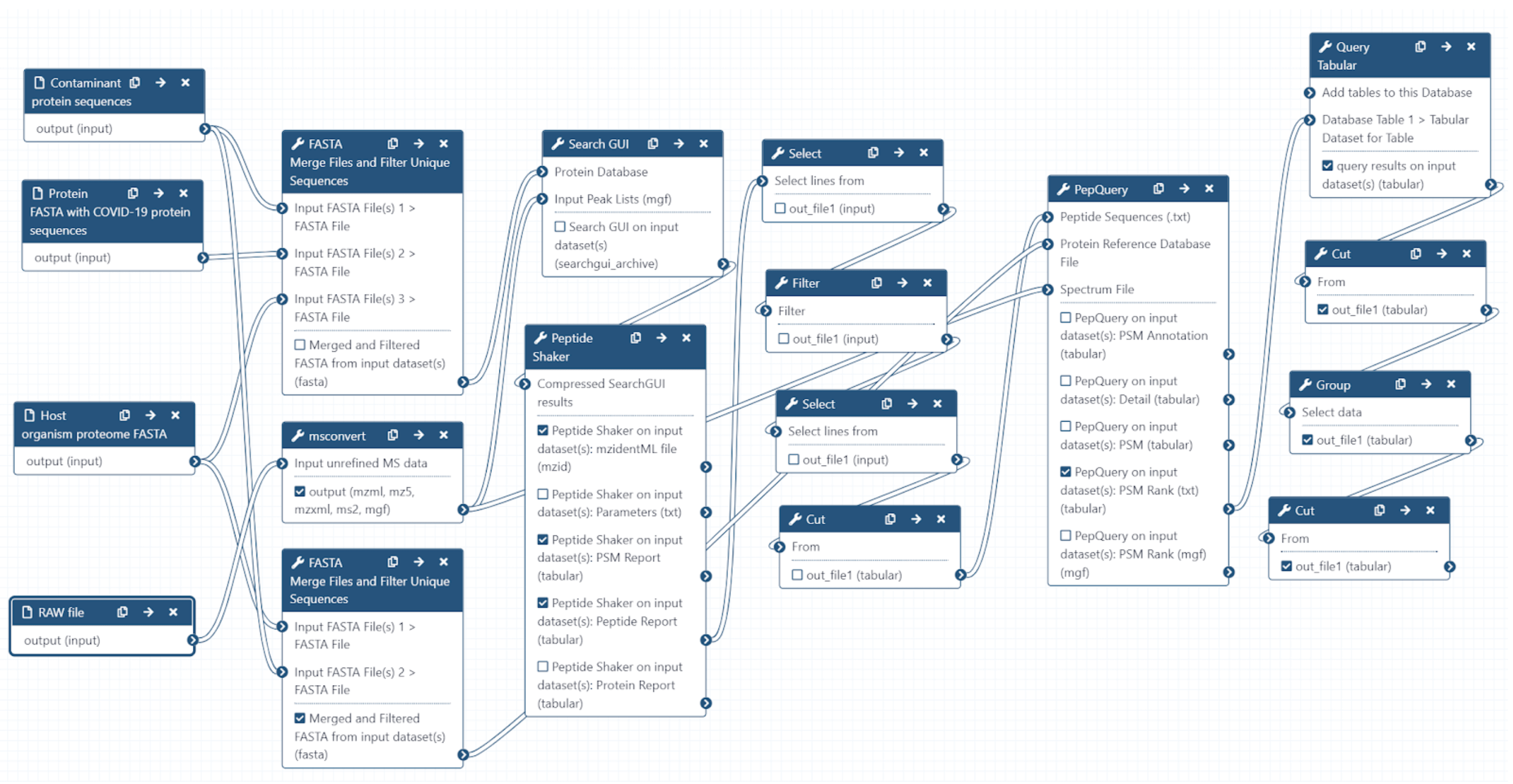
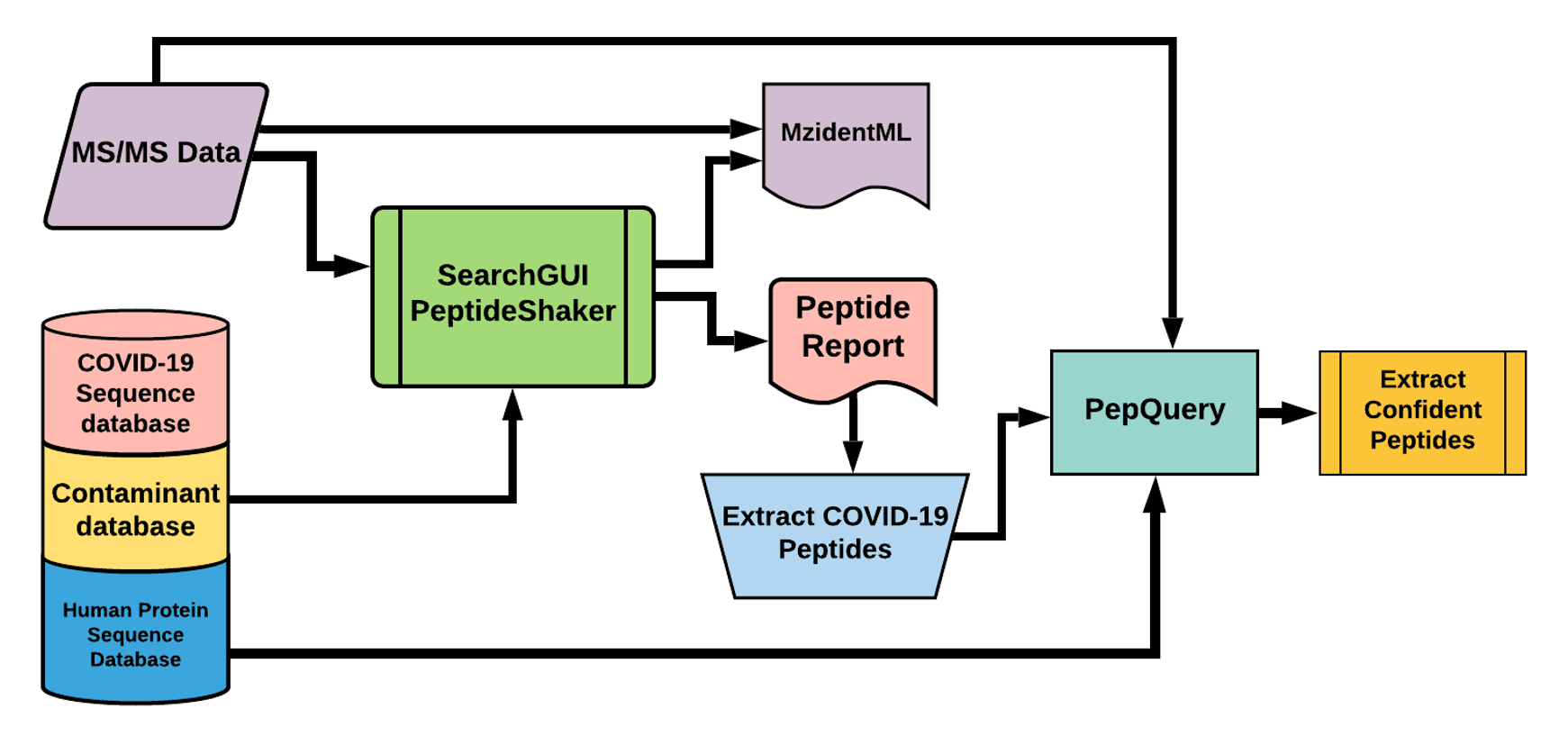
The Galaxy workflow includes RAW data conversion to MGF and mzML format. The MGF files are searched against the combined database of Human
Uniprot proteome, contaminant proteins and SARS-Cov-2 proteins database using X!tandem, MSGF+, OMSSA search algorithms with SearchGUI and FDR
and protein grouping using PeptideShaker. This resulted in detection of 579 peptides from SARS-CoV-2 proteins. The detected peptides were
searched against NCBInr to ascertain that these peptides were specific to SARS-CoV-2 proteins. The detected peptides were later subjected
to analysis by PepQuery and Lorikeet to ascertain the quality of peptide identification.
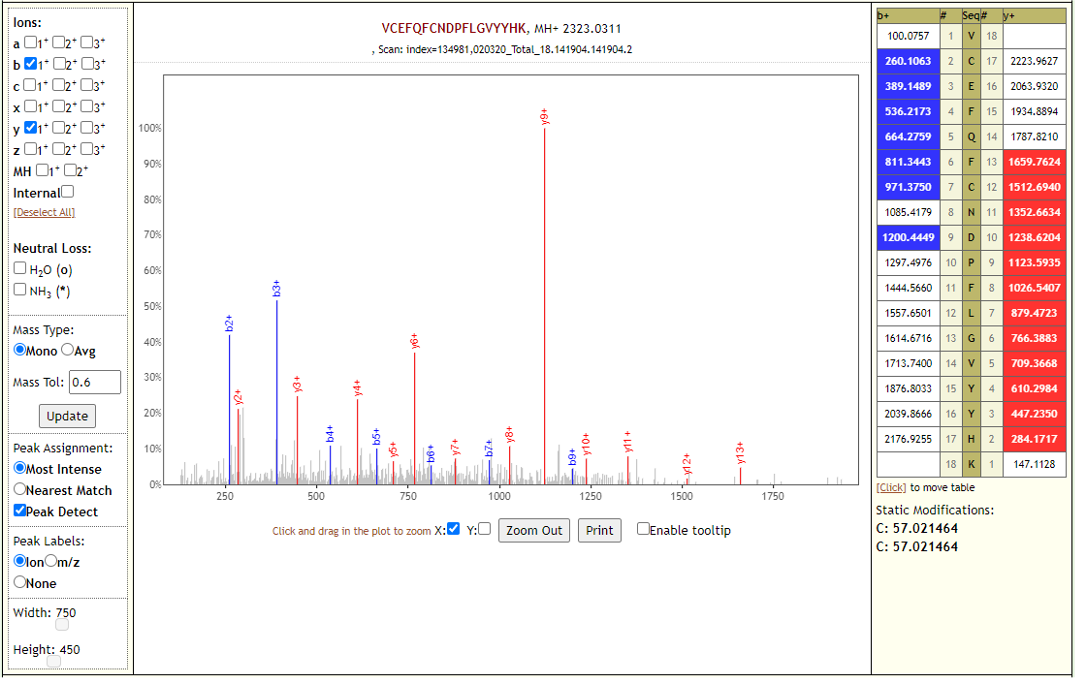
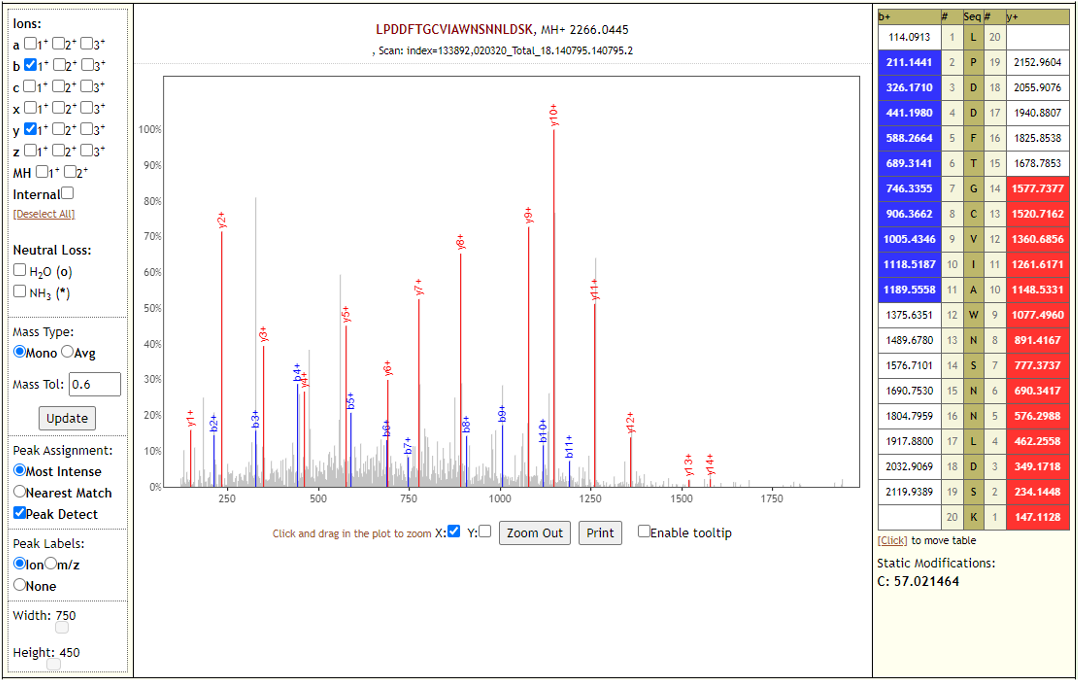
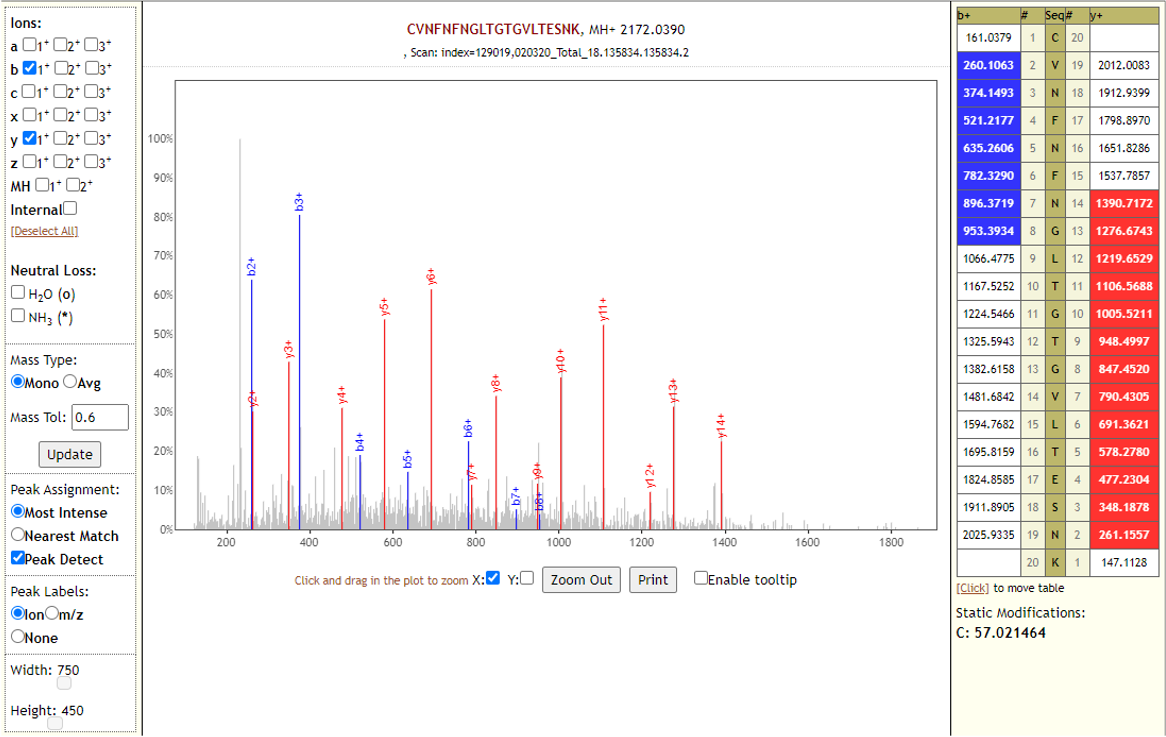
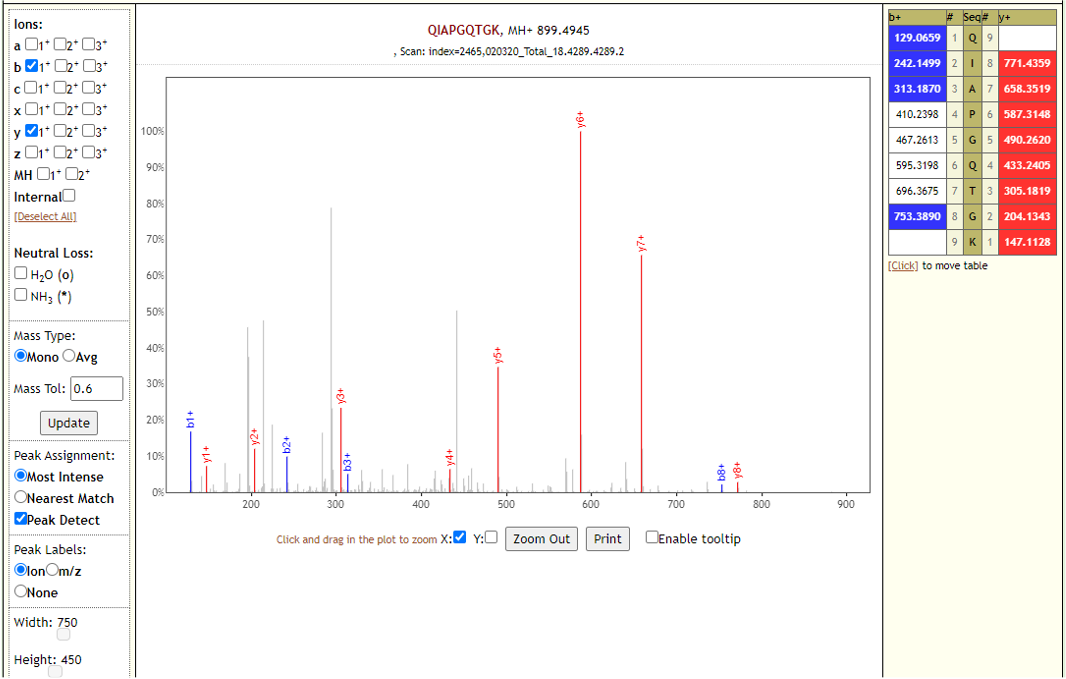
# Results
The COVID-19 positive patient samples detected 579 peptides from SARS-CoV-2 proteins. Few Lorikeet spectra of the detected peptides are shown below.