# Evolutionary Analysis
# Live Resources
| usegalaxy.org | usegalaxy.eu | usegalaxy.org.au | usegalaxy.be | usegalaxy.fr |
|---|---|---|---|---|
# What's the point?
Wu et al. (opens new window) showed recombination between COVID-19 and bat coronaviruses located within the S-gene. We want to confirm this observation and provide a publicly accessible workflow for recombination detection.
In previous coronavirus outbreaks (SARS), retrospective analyses determined that adaptive substitutions might have occurred in the S-protein Zhang et al. (opens new window), e.g., related to ACE2 receptor utilization (opens new window). While data on COVID-19 are currently limited, we investigated whether or not the lineage leading to them showed any evidence of positive diversifying selection.
# Outline
We employ a recombination detection algorithm (GARD) developed by Kosakovsky Pond et al. (opens new window) and implemented in the hyphy package. To select a representative set of S-genes we perform a blast search using the S-gene CDS from NC_045512 (opens new window) as a query against the nr database. We select coding regions corresponding to the S-gene from a number of COVID-19 genomes and original SARS isolates. This set of sequences can be found in this repository
We then generate a codon-based alignment using the workflow shown below and perform the recombination analysis using the gard tool from the hyphy package.
For selection analyses, we apply the Adaptive Branch Site Random Effects (opens new window) method to test whether or each branch of the tree shows evidence of diversifying positive selection along a fraction of sites using the absrel tool from the hyphy package.
# Inputs
A set of unaligned CDS sequences for the S-gene.
# Outputs
A recombination report:

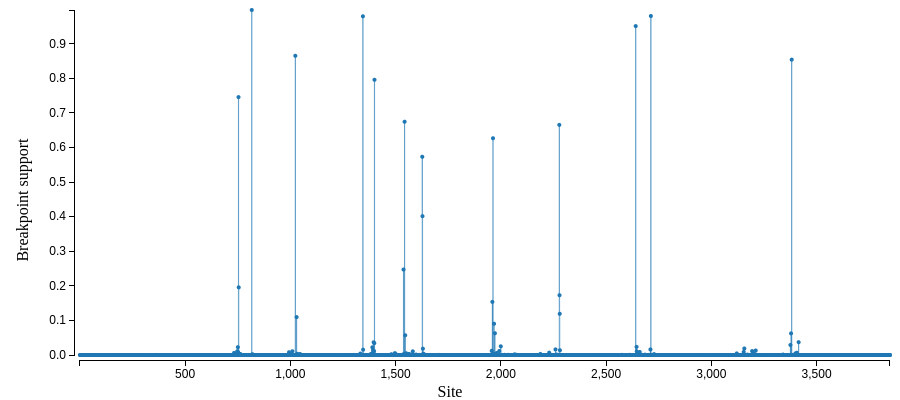
and a map of possible recombination hotspots:
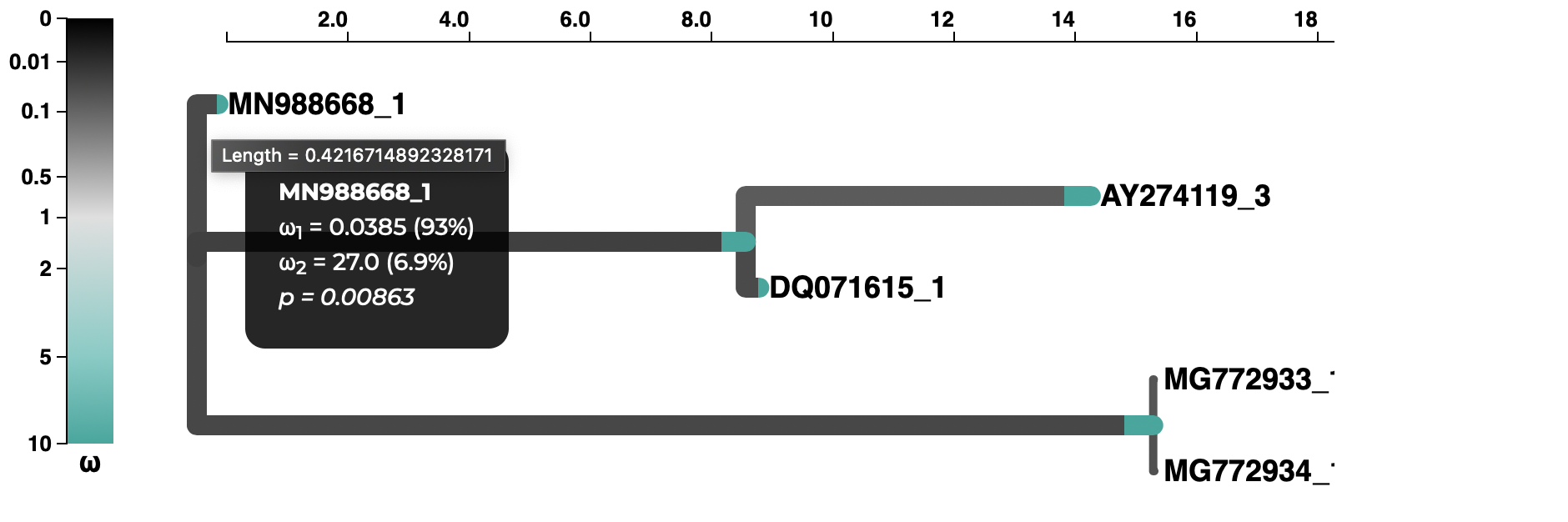
A selection analysis summary and tree (COVID-19 isolate is MN988668_1)
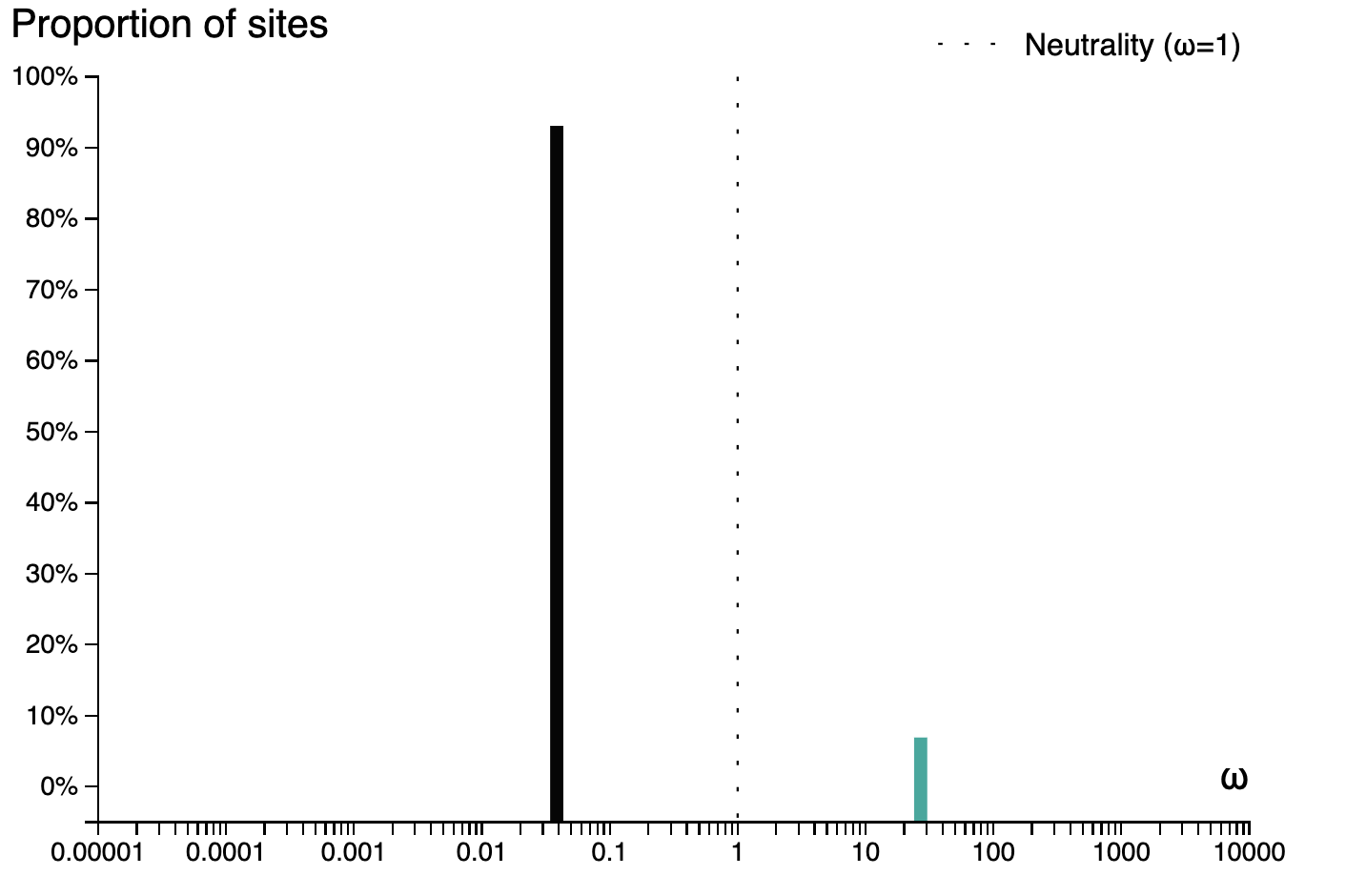
and a plot of the inferred ω distribution for the MN988668_1 branch.
# History and workflow
A Galaxy workspace (history) containing the most current analysis can be imported from here (opens new window).
The publicly accessible workflow (opens new window) can be downloaded and installed on any Galaxy instance. It contains version information for all tools used in this analysis.
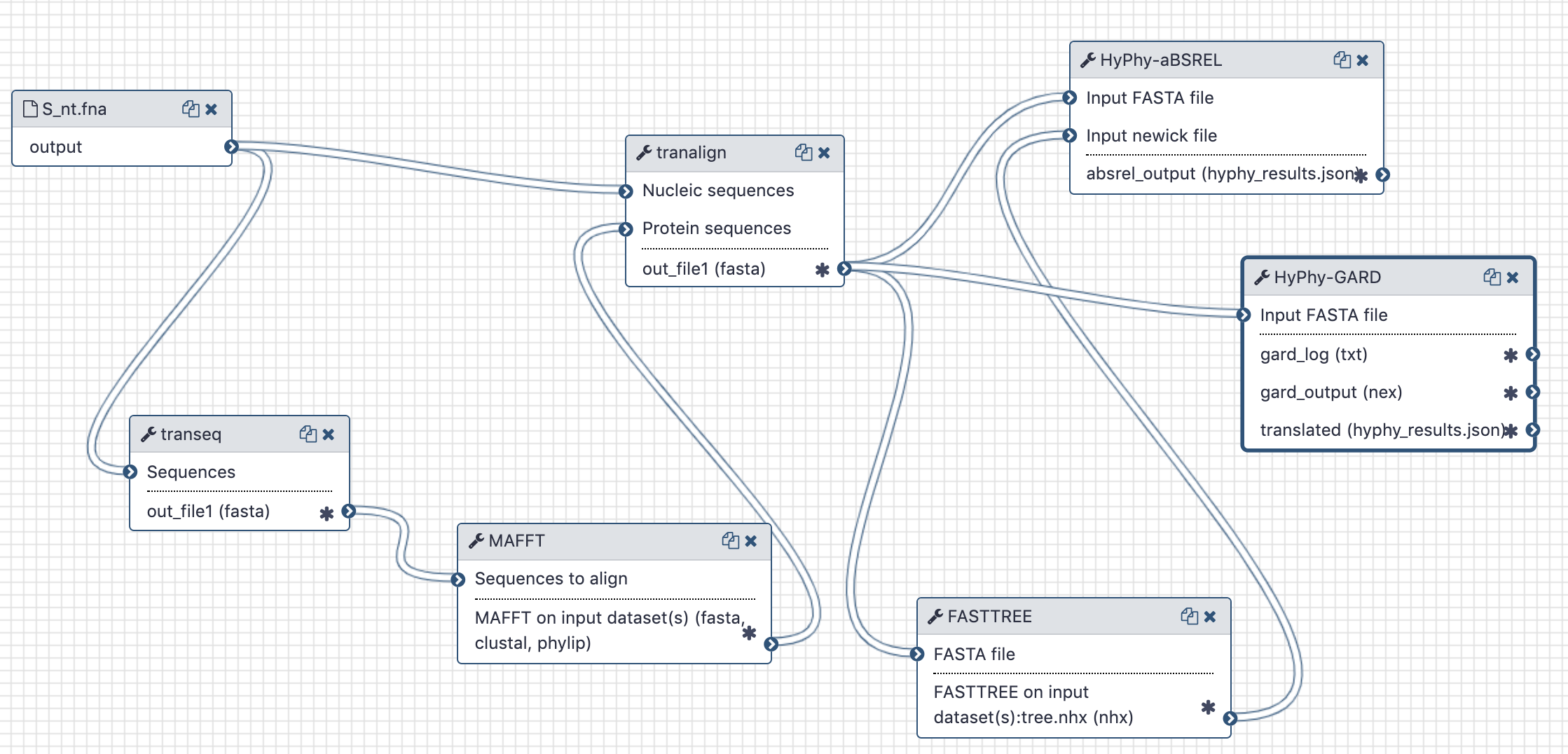
The workflow takes unaligned CDS sequences, translates them with EMBOSS:tanseq, aligns translations using mafft, realigns original CDS input using the mafft alignment as a guide and sends this codon-based alignment to gard.
# BioConda
Tools used in this analysis are also available from BioConda:
| Name | Link |
|---|---|
emboss |  (opens new window) (opens new window) |
mafft |  (opens new window) (opens new window) |
hyphy |  (opens new window) (opens new window) |
fasttree |  (opens new window) (opens new window) |