# Preprocessing of raw SARS-CoV-2 reads
The raw reads available so far are generated from bronchoalveolar lavage fluid (BALF) and are metagenomic in nature: they contain human reads, reads from potential bacterial co-infections as well as true COVID-19 reads.
# Live Resources
| usegalaxy.org | usegalaxy.eu | usegalaxy.org.au | usegalaxy.be | usegalaxy.fr |
|---|---|---|---|---|
# What's the point?
Assess quality of reads, remove adapters and remove reads mapping to human genome.
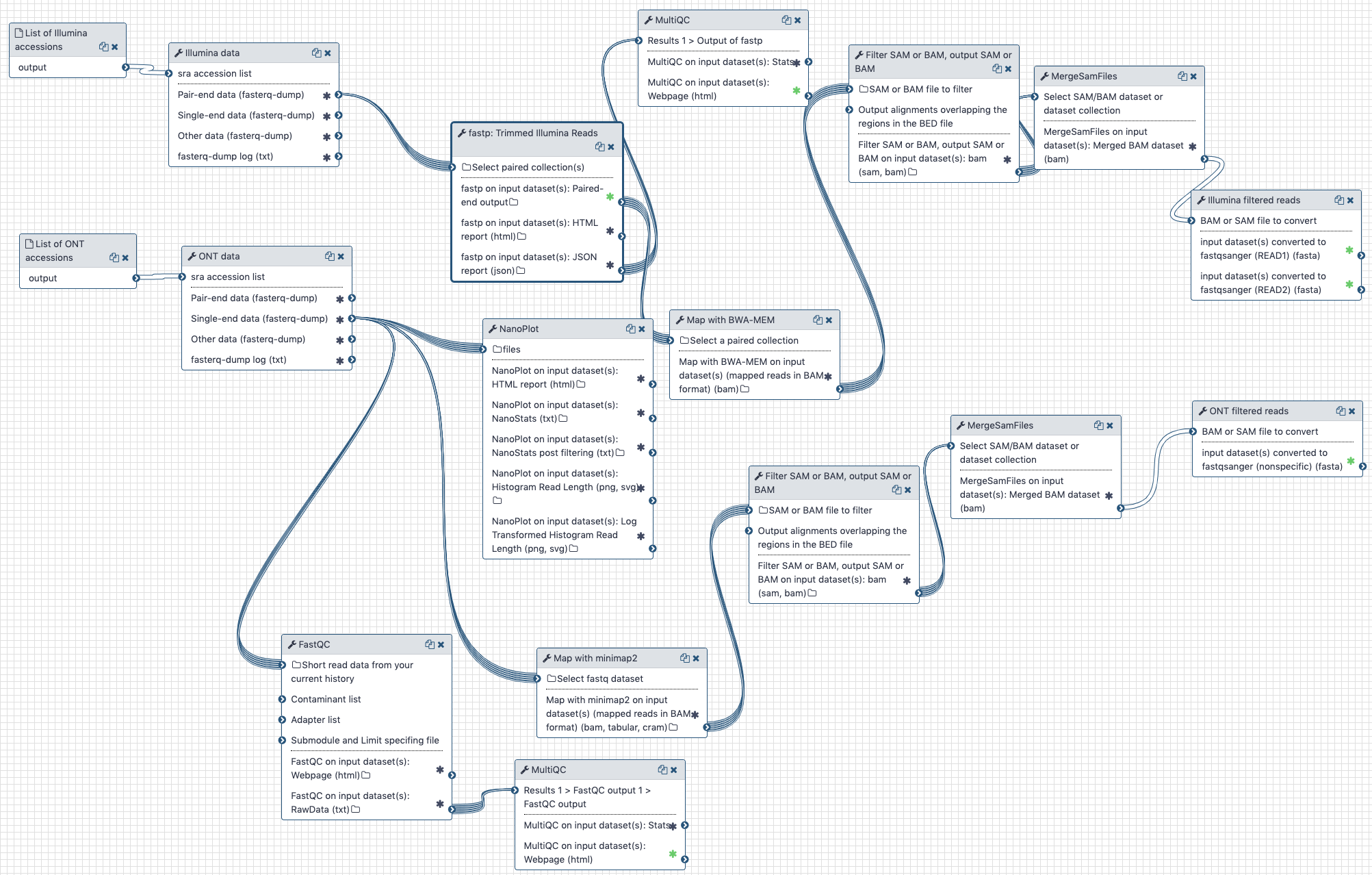
# The outline
Illumina and Oxford nanopore reads are pulled from the NCBI SRA (links to SRA accessions are available here (opens new window)). They are then processed separately as described in the workflow section.
# Inputs
💥 If you experience problems downloading data from NCBI SRA, use Galaxy history pre-populated with inputs as described in "Alternate Workflow" section below.
Only SRA accessions are required for this analysis. The described analysis was performed with all SRA SARS-CoV accessions available as of Feb 20, 2020:
Illumina reads
SRR10903401 SRR10903402 SRR10971381Oxford Nanopore reads
SRR10948550 SRR10948474 SRR10902284
# Outputs
This workflow produces three outputs that are used in two subsequent analyses:
| # | Output | Used in |
|---|---|---|
| 1. | A combined set of adapter-free Illumina reads without human contamination | Assembly |
| 2. | A combined set of Oxford Nanopore reads without human contamination | Assembly |
| 3. | A collection of adapter-free Illumina reads from which human reads have not been removed | Variation detection |
# The history and the workflow
A Galaxy workspace (history) containing the most current analysis can be imported from here (opens new window).
The publicly accessible workflow (opens new window) can be downloaded and installed on any Galaxy instance. It contains version information for all tools used in this analysis.
The workflow performs the following steps:
# Illumina
- Illumina reads are QC'ed and adapter sequences are removed using
fastp - Quality metrics are computed and visualized using
fastqcandmultiqc - Reads are mapped against human genome version
hg38usingbwa mem - Reads that do not map to
hg38are filtered out usingsamtools view - Reads are converted back to fastq format using
samtools fastx
# Oxford nanopore
- Reads are QC'ed using
nanoplot - Quality metrics are computed and visualized using
fastqcandmultiqc - Reads are mapped against human genome version
hg38usingminimap2 - Reads that do not map to
hg38are filtered out usingsamtools view - Reads are converted back to fastq format using
samtools fastx
# BioConda
Tools used in this analysis are also available from BioConda:
| Name | Link |
|---|---|
sra-tools |  (opens new window) (opens new window) |
fastqc |  (opens new window) (opens new window) |
multiqc |  (opens new window) (opens new window) |
fastp |  (opens new window) (opens new window) |
nanoplot |  (opens new window) (opens new window) |
bwa |  (opens new window) (opens new window) |
picard |  (opens new window) (opens new window) |
samtools |  (opens new window) (opens new window) |
# Alternate Workflow
An alternate starting point has been created for those not wanting to wait for the data to be downloaded from the NCBI SRA. (This can especially be an issue in Australia or Europe.)
There is a shared history (opens new window) containing all of the starting data in appropriate collections and an alternate workflow (opens new window) able to make use of this alternate input. Apart from a slightly different starting point, the workflow and the outputs it produces are identical to that above.
| usegalaxy.org | usegalaxy.eu | usegalaxy.org.au | usegalaxy.be |
|---|---|---|---|