# Docking
This section describes the docking procedure. This was repeated 17 times, for each of the 17 fragment screening crystal structures that were available at the time (more are expected).
# Live Resources
| usegalaxy.eu |
|---|
# Outline
Docking is performed with rDock [1] using as inputs:
- PDB file of the protein of the fragment screening crystal structure with the ligand and waters removed, and protonated (using OpenBabel [2]) at pH 7.4
- The active site definition for that protein prepared as described in step 2
- Candidate molecules prepared as described in step 1
25 poses were generated for each molecule.
# Inputs
A complete list of all inputs is provided in this history (opens new window). The hits_frankenstein_17.sdf file contains the 'Frankenstein ligand' used to generate the active site; the Mpro-x*as files contain active site definitions for all 17 fragments; the hits.sdf file contains the structures of the fragment hits; and the EnumeratedCandidates collection contains all candidates prepared for docking.
# History and workflow
A Galaxy workspace (history) containing the most current analysis can be imported from here (opens new window).
The publicly accessible workflow (opens new window) can be downloaded and installed on any Galaxy instance. It contains version information for all tools used in this analysis.
| Docking |
|---|
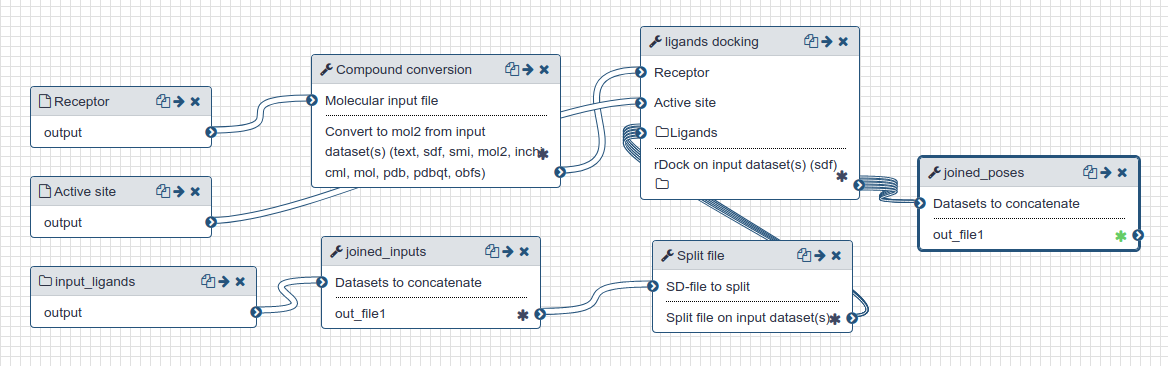 |
| The docking workflow. |
# References
[1] Ruiz-Carmona et al. (2014). rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Computational Biology. 10 (4). doi:10.1371/journal.pcbi.1003571 (opens new window).
[2] Open Babel: An open chemical toolbox. O'Boyle et al. doi:10.1186/1758-2946-3-33 (opens new window).